


How can I view RNASeq data in Ensembl?
RNASeq data are available for more than 20 species in Ensembl. These data have been processed using our in-house Ensembl RNASeq pipeline. For most species, we include the following data:
- BAM files, which can be viewed on our website or downloaded from our FTP site. Click on the peaks to access more information about the read alignment. Zoom in to see which bases in each read match (grey) or don't match (red) the BAM consensus sequence. Reads with a green edge are paired-end reads that have aligned nearby their mate. Green edges indicate a level of confidence that a read has aligned to the correct region of the genome. Reads with a red edge are paired-end reads that have a mate that did not align nearby or did not align at all to the genome. Red edges indicate a level of uncertainty in the alignment. Reads with a blue edge are single-end reads that did not have a mate.
- Introns (splice junctions) identified by reads that splice when they align to the genome. The height of each intron block is indicative of the the number of reads found to splice across the intron. Click on an intron block to see its exact genomic coordinates and the number of reads that support it.
- Gene models constructed using RNASeq data.
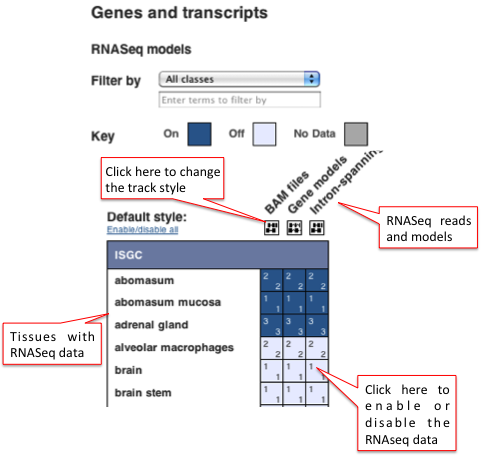
These three types of RNASeq data can be viewed alongside the genome in the Location or Gene tabs by clicking on 'Configure this page' . In the configuration window, under the 'Genes and transcripts' menu on the left hand side, click on 'RNASeq models'.
You can filter by tissue type, e.g. blood, and in human, you can filter by classes of data, e.g. Human BodyMap 2.0 and Beta cell transcriptome. You can choose to view the BAM files, the Gene models and/or the Intron-spanning reads for specific tissues using the configuration matrix. Watch this video on the Ensembl Regulation data to see how to turn tracks on and off using any configuration matrix. See the images below for an example in sheep.

If you have any other questions about Ensembl, please do not hesitate to contact our HelpDesk. You may also like to subscribe to the developers' mailing list.