


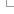
Ensembl Bacteria pipeline
Ensembl Bacteria contains genomes from annotated INSDC records that are loaded into Ensembl multi-species databases, using the INSDC annotation import pipeline. This document describes how this pipeline is used for bulk loading of bacteria and provides tips for handling the data.
Identifying genomes
Ensembl Bacteria consists of completely sequenced genomes from eubacteria and archaea, which sequences have been deposited in INSDC. We then query the ENA Genomes Assembly Database to find assembly sets for eubacteria and archaea which have at least 50 CDS annotations. Assembly sets are grouped into collections of up to 250 genomes. There are no strict rules guiding the assignment of genomes to particular collections, but a given genome will usually stay in the same collection. The collections of genomes are then passed to the INSDC annotation import pipeline for loading into Ensembl.
Genome names and identifiers
Genomes in Ensembl Bacteria are assigned the names and NCBI taxonomy identifiers obtained from the corresponding ENA Genomes Assembly Database record. However these names and identifiers are not always unique, and exact names may change between releases. For some genomes, the most stable identifiers can be the unique assembly accessions assigned by the ENA Genomes Assembly Database, e.g. GCA_000005845.1. Each assembly also has an externally assigned name, also obtained from the ENA Genome Assembly Database, e.g. ASM584v1. This may be used to disambiguate cases where the organism names happen to be the same (e.g. Borrelia afzelii PKo (ASM22283v1) vs. Borrelia afzelii PKo (ASM16559v2)).
Dealing with identifier problems
For older records (and other special cases), locus_tag and/or protein_id qualifiers can be either missing or duplicated within/between records. This may reflect errors in annotation or submission, and systematic problems such as the reuse of locus tag prefixes between different entries. We try to resolve identifiers problems as follows:
- intra-genome problems
- collate all features with duplicated identifiers and
- reject genomes with features of the same types with the same identifier or
- if duplicates are shared between features of different types, retain for protein_coding and discard for others
- collate all features with missing identifiers
- protein_coding genes
- if
- otherwise, reject the genome
- for all other missing identifiers, a new Ensembl identifier of the form ENA[GTP]n (e.g. ENAG000001) is assigned (this is stable provided the underlying feature does not change)
- protein_coding genes
- collate all features with duplicated identifiers and
- inter-genome problems
- find all sets of genomes that share duplicated identifiers
- retain identifiers on the genome that was submitted first
- discard the genome if the level of duplication is less than 50% (likely to be individual annotation/submission errors)
- assign new Ensembl identifiers if the duplication is more than 50% (likely to be a systematic error such as prefix reuse)
Other data sources
In addition to the annotation loaded using INSDC, the following data sources are also used to enrich the annotation of our genomes:
- Data from RegulonDB is used to add polycistronic transcripts, and operons and other regulatory features, to the Escherichia coli K-12 MG1655 reference genome
- Rhea and MetaCyc cross-references are added using data from Microme
Comparative Genomics
Owing to the number of genomes included in Ensembl Bacteria, gene trees and whole genome aligments are not calculated for all genomes, but gene families are still populated based on InterPro annotation. A large number of bacterial genomes are included in the Ensembl Genomes pan-taxonomic compara for which gene trees are built using selected genomes from across the taxonomy.
![[twitter logo]](/i/twitter.png "Follow us on Twitter!")
